Current projects
Deciphering the role of host genetics in the pathogenesis and suceptibility to severe COVID-19
Deciphering the role of host genetics in the pathogenesis and suceptibility to severe COVID-19
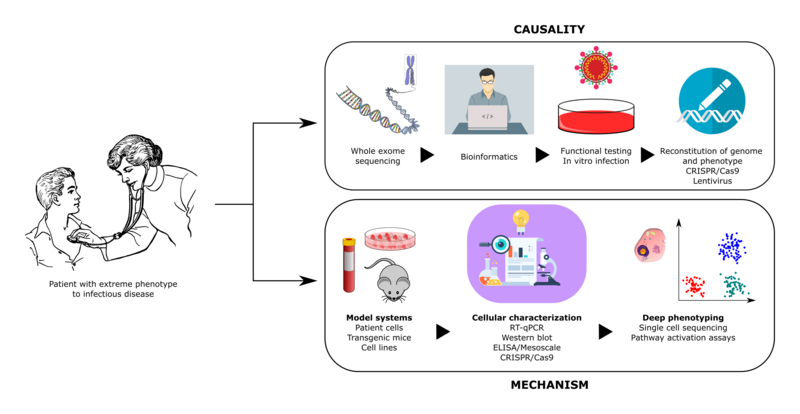
During the current COVID-19 pandemic, stunning inter-individual variability among infected individuals, ranging from asymptomatic infection to lethal COVID-19, has been observed. In this context, we hypothesize that life-threatening COVID-19 in individuals without co-morbidities can be caused by rare defects in genes with essential roles in antiviral immune responses to SARS-CoV2. To test this hypothesis, our project includes the following aims: 1) recruitment of otherwise healthy patients with severe COVID-19 within an international covidhge consortium 2) whole-exome sequencing to search for disease-causing gene variants, 3) functional studies including SARS-CoV2 infection to characterize immune responses in patient cells, and 4) translation of this knowledge into the clinic to develop genetic tests and plasma assays to identify healthy individuals and patients at increased risk of developing severe COVID-19. Thereby we hope to gain a better understanding of disease mechanisms to ensure implementation of prophylactic measures or intensified treatment modalities to prevent severe disease and improve disease outcome in patients with COVID-19.
Role of autophagy in the antiviral defence against herpesviruses
Role of autophagy in the antiviral defence against herpesviruses
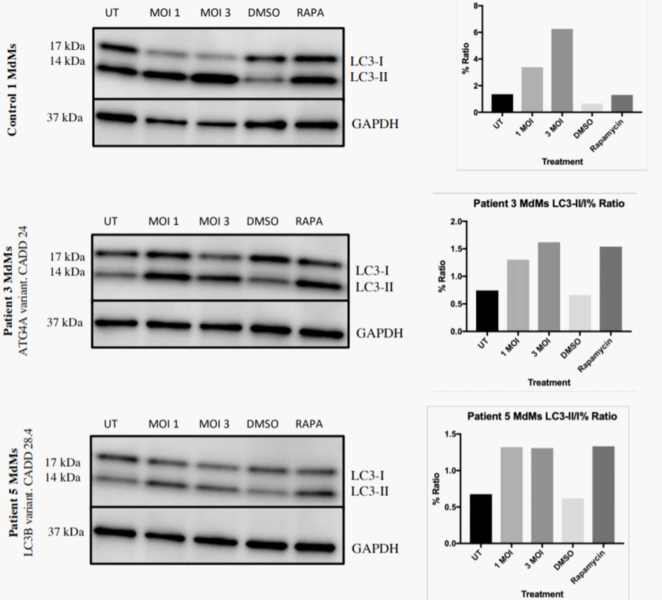
The immune system is essential for the control of infections, but also causes significant inflammation and tissue damage in doing so. Some organs, most notably the brain, are very susceptible to this immunopathology. We therefore hypothesize that more "silent" immune protecting mechanisms exist and constitute a first line of defense in these areas. These innate systems are largely unknown, and there is specifically no knowledge on how the human brain fights infections independent of classical immunological activities. In this proposal, we hypothesize the cell-autonomous mechanism autophagy to be a potent anti-viral mechanism, controlling viral replication in neurons without activating inflammatory responses. We have whole-exome-sequenced 18 patients with recurrent HSV2 (Mollaret’s) meningitis and found that 8 of them have predicted loss-of-function mutations in autophagy genes. In this project, we will thus explore the role of autophagy in antiviral defense in general, and specifically whether the identified mutations are responsible for elevated susceptibility to virus infections in neurons. For this purpose, we will use a broad panel of frontline methods including whole exome sequencing on clinically well-defined patients, stem cell-derived neurons, and CRISPR/Cas9 technology. Collectively, this could lead to the identification of novel modes of innate host protection, that can fight infections while sparing healthy tissue.
Identification of novel innate immunodeficiencies in patients with varicella-zoster virus encephalitis
Identification of novel innate immunodeficiencies in patients with varicella-zoster virus encephalitis

Varicella-zoster virus (VZV) is a common virus and is part of the herpesvirus family. The first infection with VZV causes chickenpox, whereafter the virus has the ability to remain inactive in the sensory nerves. The virus can later reactivate thereby causing shingles in a belt-like area of the skin. A rare but serious complication to VZV infection is the risk of the virus spreading through the nerves to the central nervous system and the brain. This results in the development of encephalitis, an acute inflammation of the brain. Only limited treatment against encephalitis caused by VZV exists, and the virus often leaves patients with permanent injuries.
We assume that some patients have alterations in their genes (mutations) that might increase their susceptibility to virus infections in the brain. Especially alterations in genes important for our immune response system, more specifically the production of specific molecules called “Type 1 Interferons”.
Our first aim is to identify these exact alterations (mutations) in patient genes. We have access to all adult patients in Denmark who have suffered from VZV encephalitis since 2014, and we will collect blood samples from all patients enrolled in the study. By using the technique “whole exome sequencing” it is thus possible to investigate the patients’ genes to identify mutations. We will afterwards investigate if these mutations alter/decrease the function of the immune response system by performing different tests with both patient cells and “healthy” control cells. This knowledge will provide a greater insight into the VZV disease progression and will be valuable in the future development of improved patient diagnosis, treatment and prevention.
Genetic and immunological determinant of severe influenza virus infection in humans
Genetic and immunological determinant of severe influenza virus infection in humans
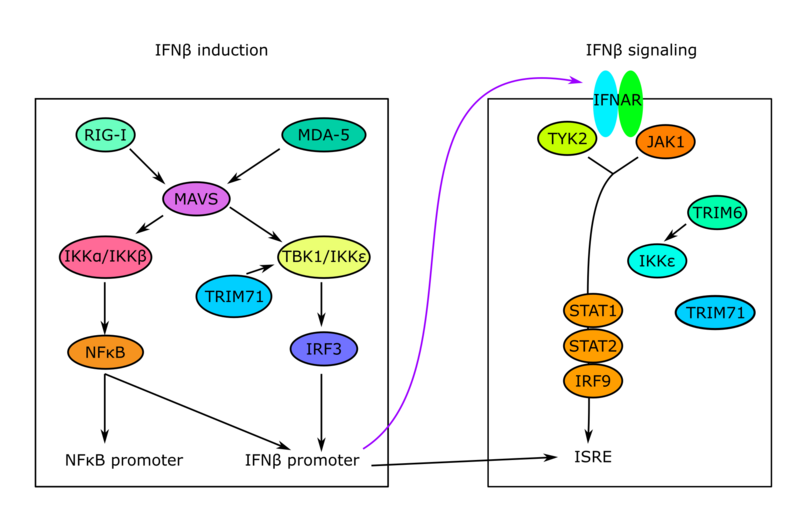
Influenza infection is common worldwide with many individuals affected during epidemics and pandemics. Previous studies have established an essential role for innate immune responses, and in particular the production of Type I and III interferon (IFN), for viral elimination and survival from influenza infection in vitro and in animal models. However, genetic and immunological determinants of very severe disseminated influenza infection in humans remain poorly characterized. In 2015, the first report of a primary immunodeficiency involving IFN regulatory factor (IRF)7 in a child with severe disseminated influenza was published. Subsequently our group and others have described defects in innate receptors of cytosolic dsRNA, including TLR3 and RIG-I, as well as defective signaling by the transcription factors IRF3, IRF7 conferring susceptibility to severe influenza. In this project, we will collect a cohort of young adult patients admitted to the intensive care unit (ICU) with severe influenza A virus (IAV) H1N1 and perform whole exome sequencing (WES) to identify potentially disease-causing genetic variants. This will be followed by functional immunological and virological analyses to examine innate antiviral immune responses in relevant cell populations from patients. The impact of variants will also be investigated in HEK 293T cells made devoid of the molecule of interest to compare IFNβ-reporter activity when transfecting wild-type (WT) and mutant protein-encoding plasmids into cells. Finally, we will perform reconstitution experiments on patient cells to demonstrate reversion to the normal phenotype by the introduction of the WT protein in question. This is aiming at establishing a direct causal association between the gene variant identified by WES, the immunological phenotype of patient cells and the clinical presentation of the patient.
Overall the present project has the potential to contribute with important new insights into the pathogenesis of influenza infection in humans. Importantly, such knowledge may have clinical perspectives and pave the way for improved prophylaxis and treatment of patients with severe influenza